Terapie target anti-EGFR nel tumore del polmone: Razionale biologico e meccanismi molecolari dei TKI
Terapie target anti-EGFR nel tumore del polmone: Razionale biologico e meccanismi molecolari dei TKI (Tyrosine Kinase Inhibitors)
Introduzione
La comprensione dei meccanismi molecolari alla base dello sviluppo del tumore al polmone ha profondamente modificato l’approccio terapeutico a questa patologia. In particolare, l’identificazione di specifiche alterazioni genetiche driver, capaci di guidare la crescita tumorale, ha permesso lo sviluppo di terapie mirate, dette terapie target.
Tra queste, un ruolo centrale è svolto dagli inibitori della tirosin-chinasi (TKI) diretti contro il recettore EGFR (Epidermal Growth Factor Receptor). Questi farmaci rappresentano uno degli esempi più emblematici di come la biologia molecolare possa tradursi direttamente in applicazione clinica, consentendo un intervento selettivo sui meccanismi che sostengono la proliferazione tumorale.
Il recettore EGFR: struttura e funzione biologica
EGFR è un recettore tirosin-chinasico transmembrana, appartenente alla famiglia ErbB. È costituito da:
• un dominio extracellulare, deputato al legame con il ligando
• un dominio transmembrana
• un dominio intracellulare tirosin-chinasico, responsabile dell’attività catalitica
In condizioni fisiologiche, il legame del ligando induce la dimerizzazione del recettore, evento che attiva la funzione chinasica e avvia la fosforilazione di residui tirosinici su specifiche proteine bersaglio.
Questa cascata di segnali regola processi fondamentali quali:
• proliferazione cellulare
• differenziamento
• migrazione
• sopravvivenza
Nel tumore al polmone non a piccole cellule (NSCLC), mutazioni attivanti del gene EGFR determinano una attivazione costitutiva del recettore, indipendente dal legame con il ligando, producendo uno stimolo proliferativo continuo.
Le mutazioni più comuni riguardano:
• delezioni dell’esone 19
• mutazione puntiforme L858R dell’esone 21
Queste alterazioni determinano un cambiamento conformazionale del dominio tirosin-chinasico, aumentando la sua affinità per l’ATP e potenziando la trasduzione del segnale.
Meccanismo molecolare d’azione dei TKI
Competizione con l’ATP
Il dominio tirosin-chinasico di EGFR utilizza ATP come donatore di gruppi fosfato per la fosforilazione delle proteine intracellulari. I TKI sono molecole di piccole dimensioni progettate per occupare il sito catalitico dell’ATP, impedendone il legame.
Questo meccanismo, noto come inibizione competitiva, blocca l’attività enzimatica del recettore e arresta l’intera cascata di segnali intracellulari.
In termini molecolari, i TKI stabiliscono interazioni specifiche con:
• residui amminoacidici chiave del dominio chinasico
• la tasca di legame dell’ATP
• regioni regolatorie che controllano la conformazione attiva del recettore
Blocco della fosforilazione e delle vie di segnale intracellulari
L’inibizione dell’attività chinasica di EGFR determina la soppressione di numerose vie di segnalazione oncogeniche, tra cui:
• PI3K–AKT–mTOR, coinvolta nella sopravvivenza cellulare e nella resistenza all’apoptosi
• RAS–RAF–MEK–ERK, principale asse mitogenico responsabile della proliferazione
• JAK–STAT, implicata nella regolazione dell’espressione genica
Il blocco simultaneo di queste vie produce:
• arresto del ciclo cellulare
• riduzione della sintesi proteica
• attivazione dei meccanismi apoptotici
Questo spiega l’elevata efficacia dei TKI nei tumori EGFR-dipendenti, ovvero neoplasie la cui crescita è fortemente guidata dall’iperattivazione di questo recettore (Fig.1).
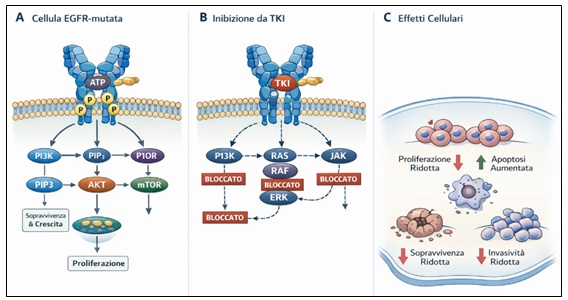
Fig.1: Meccanismo d’azione degli EGFR-TKI nel tumore del polmone con mutazione di EGFR
Classificazione dei TKI: evoluzione molecolare e farmacologica
Nel corso degli anni sono stati sviluppati TKI sempre più selettivi ed efficaci, suddivisi in tre generazioni.
TKI di prima generazione
Gefitinib ed Erlotinib sono inibitori reversibili che competono con l’ATP per il sito catalitico del recettore.
Caratteristiche principali:
• elevata affinità per le forme mutate di EGFR
• selettività discreta rispetto alla forma wild-type
• rapida risposta clinica iniziale
Il limite principale è rappresentato dallo sviluppo di resistenza acquisita, frequentemente dovuta alla mutazione T790M, che altera la conformazione del sito di legame rendendo inefficace l’interazione con il farmaco.
TKI di seconda generazione
Afatinib e Dacomitinib sono inibitori irreversibili, capaci di formare legami covalenti con residui specifici del dominio tirosin-chinasico.
Questa modalità d’azione determina:
• maggiore stabilità dell’inibizione
• blocco prolungato del segnale
Essi presentano inoltre un’attività più ampia verso altri membri della famiglia ErbB, contribuendo a un controllo più completo della rete di segnalazione oncogenica.
TKI di terza generazione
Osimertinib rappresenta il più avanzato sviluppo della progettazione molecolare dei TKI.
È stato specificamente progettato per:
• inibire le mutazioni classiche
• superare la resistenza mediata dalla mutazione T790M
• minimizzare l’attività sul recettore wild-type
Dal punto di vista strutturale, Osimertinib presenta una configurazione molecolare altamente selettiva, capace di discriminare finemente tra EGFR mutato e non mutato, garantendo un’inibizione altamente mirata.
Selettività biologica delle terapie target
A differenza della chemioterapia tradizionale, che colpisce indiscriminatamente tutte le cellule in proliferazione, i TKI esercitano la loro attività solo sulle cellule che esprimono la specifica alterazione molecolare bersaglio.
Questo principio, noto come oncogene addiction, descrive la dipendenza critica della cellula tumorale da un singolo pathway molecolare iperattivato.
Nei tumori EGFR-mutati:
• l’inibizione di EGFR provoca un collasso funzionale dell’intera rete di segnale
• le cellule sane, che non dipendono in modo esclusivo da questo recettore, risultano relativamente risparmiate
Questo spiega l’elevato indice terapeutico dei TKI.
Collegamento biologico–clinico: la dipendenza oncogenica
Il concetto di dipendenza oncogenica rappresenta il fondamento biologico della terapia target.
Le cellule tumorali EGFR-mutanti sviluppano una forte dipendenza funzionale dall’iperattivazione del recettore. Questo comporta:
• perdita della plasticità dei pathway di segnale
• ridotta capacità di attivare vie alternative
• vulnerabilità selettiva all’inibizione farmacologica
Dal punto di vista clinico, ciò si traduce in:
• elevata percentuale di risposta
• rapida riduzione del carico tumorale
• controllo prolungato della malattia
Ruolo centrale della diagnostica molecolare
L’efficacia dei TKI è strettamente subordinata alla corretta identificazione delle mutazioni di EGFR. Questo rende fondamentale l’esecuzione di:
• analisi molecolari su tessuto tumorale
• biopsia liquida mediante DNA tumorale circolante
L’integrazione tra biologia molecolare e clinica permette di selezionare con precisione i pazienti candidabili alla terapia, configurando un modello di medicina di precisione.
Conclusioni
Le terapie target anti-EGFR rappresentano una delle applicazioni più avanzate della biologia molecolare traslazionale. Attraverso la comprensione dettagliata dei meccanismi di segnalazione cellulare e delle alterazioni genetiche che guidano la crescita tumorale, è stato possibile sviluppare farmaci capaci di colpire selettivamente il bersaglio molecolare.
I TKI non costituiscono semplicemente una nuova classe farmacologica, ma rappresentano un vero cambio di paradigma terapeutico, basato sulla conoscenza profonda della biologia del tumore.

Dott. ssa Jessica Evangelista
Policlinico Universitario Agostino Gemelli – Roma
